 Hội Nội Tiết – Đái Tháo Đường Miền Trung Việt Nam Hội Nội Tiết – Đái Tháo Đường Miền Trung Việt Nam
Hội Nội Tiết – Đái Tháo Đường Miền Trung Việt Nam Hội Nội Tiết – Đái Tháo Đường Miền Trung Việt Nam
TRÍ NHỚ CHUYỂN HÓA VÀ CÁC BIẾN CHỨNG BỆNH ĐÁI THÁO ĐƯỜNG
GS.TS. Thái Hồng Quang
SUMMARY
Metabolic memory and diabetic complications
Many important biochemical mechanisms are activated in the presence of high levels of glucose, which occur in diabetes. Large randomised studies have established that early intensive glycaemic control reduces the risk of diabetic complications. Environmental factors, such as diet and exposure to hyperglycemia, contribute to the etiology of diabetes mellitus and its associated microvascular and macrovascular complications. These vascular complications are the main cause of the morbidity and mortality burden of diabetes mellitus.
The DCCT–EDIC and UKPDS epidemiological studies correlated poor glycemic control with the development of vascular complications in patients with type 1 or type 2 diabetes mellitus. The findings of these studies suggest that early exposure to hyperglycemia predisposes individuals to the development of diabetic complications, a phenomenon referred to as metabolic memory or the legacy effect. The first experimental evidence for metabolic memory was reported >20 years ago and the underlying molecular mechanisms are currently being characterized. Interestingly, transient exposure to hyperglycemia results in long-lasting epigenetic modifications that lead to changes in chromatin structure and gene expression, which mediate these persistent metabolic characteristics.
Chịu trách nhiệm chính: Thái Hồng Quang
Ngày nhận bài: 7.6.2016
Ngày phản biện khoa học: 23.6.2016
Ngày duyệt bài: 1.7.2016
Glucose gây độc hại ở các tổ chức khác nhau (nhiễm độc glucose) đã được biết đến từrất lâu.Thực nghiệmtrên mô hình động vật đã được tiến hành trước đây, giữa những năm 1980 đã ra đời khái niệm “trí nhớ chuyển hóa”. Giữa những năm 2000 – 2002, các kết quả nghiên cứu lâm sàng đã chứng minh rằng, điều trị sớm, tích cực để kiểm soát đưa glucose trở về bình thường có thể làm giảm tiến triển xấu chuyển hóa trong tế bào và hạn chế tới mức thấp nhất các nguy cơ các biến chứng mạn tính (1,2).
Nhiều nghiên cứu ở nhiều trung tâm khác nhau sau đó đã xác nhận kết quả của những nghiên cứu này (6). Các biến chứng mạn tính là kết quả do tác động của một số tác nhân như: chuyển hóa, hormone, môi trường
hoặc gene.
Tăng glucose máu kéo dài là nguyên nhân gây nên các biến chứng bệnh lý vi mạchmạn tính (bệnh võng mạc, bệnh thận, bệnh thần kinh), cũng như bệnh mạch máu lớn (bệnh tim thiếu máu cục bộ, bệnh mạch máu não và bệnh mạch máu ngoại vi).
Tình trạng tăng glucose mạn tính hoạt hóa một số chuyển hóa: Glycat hóa protein không enzyme (non-enzymatic protein glycation), Chuyển hóa polyol(polyol pathway) và stress oxy hóa. Tăng nồng độ glucose làm biến đổi thành phần LDL, chính LDL sẽ gây nên nhiễm độc nó trong nội mạc.
Tăngglucose mạn tính cùng với với tình trạng viêm sẽ làm rối loạn cân bằng giữa metalloproteinase với các yếu tố ức chế nó (MMP/TIMP) gây nên tái cấu trúc bệnh lý thành mạch máu, tăng sinh nội mạc và tân tạo mạch (arteriogenesis). Gần đây đangthảo luận về khái niệm “trí nhớ tăng glucose máu”, một hiện tượng của quá trình bệnh lý kéo dài liên quan với tăng stress oxy hóa, glycat hóa protein và lipid của tế bào.
Hiện tượng này khởi đầu như là hậu quả của tăng glucose máu ngay lúc bắt đầu khởi phát bệnh. Trí nhớ của các cơ quan và tổ chức sẽ ghi nhớ tình trạng tăng glucose ngay lúc ban đầu này, mặc dầu sau đó kiểm soát chuyển hóa đã được cải thiện (3).
Tăng glucose máu nhất thời (transient) hoạt hóa những thay đổi epigenetics (biểu sinh) trong thời gian dài ở vùng khởi động (promoter) của NFkB tiểu đơn vị 65. Những tác dụng epigenic kéo dài muộn về sau có sự trung gian bởi tập hợp các enzymes tham gia tái biến đổi histone (histone modifying enzymes), các enzymes này tái cấu trúc các sợi chromatin DNA, nhờ vậy quá trình phiên mã trong gene xẩy ra. (Turner – 2007).
Từ đó, chủ trương điêu trị tích cực, cẩn thận bệnh đái tháo đường ngay từ thời điểm chẩn đoán bệnh không phải là vấn đề cường điệu. Đây là vấn đề đặc biệt, giai đoạn sớm của bệnh, theo một mức độđáng phải lưu ý, là yếu tố đi trước của những biến chứng mạn tính sau này, như đây là số mệnh của người bệnh vậy, vìtăng glucose mạn tính sẽ kích hoạt nhiều quá trình bệnh lý bất lợi như:
- Hoạt hóa protein kinase C và phospholipase A2 làm tăng các sản phẩm chuyển hóa arachidonic acid.
- Tăng bộc lộ các yếu tố phát triển làm tăng co mạch (vasoconstriction)
- Tăng AGEs, tăng stress oxy hóa thông qua việc gắn với các receptors AGE thích hợp (RAGE)
- Glycat hóa và oxy hóa LDL.
- Tăng bộc lộ yếu tố phiên mã nhân NFkβ, tăng nồng độ các phân tử kêt dính, giảm sinh khả dụng NO, tăng IL-6, TNF-α, và các cytokine khác.
- Hoạt hóa protein kinaseC và tăng DAG (diacyloglycerol) đưa đến giảm tổng số NADPH và tạo NO.
Nhiều công trình nghiên cứu hệ thống đã phát hiện quá trình bệnh lý cơ bản của tất cả các cơ chế bệnh lý này: Sản xuất quá mức free oxygen radical superoxide và reactive oxygen species (ROS) là nguyên nhân đưa đến các các rối loạn chuyển hóa trên. Superoxide hoạt hóa các đường bệnh lý chủ yếu do tăng glucose máu do giảm hoạt tính của enzyme chìa khóa trong quá trình ly giải glucose (glycolytic enzyme) – glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Superoxide được sinh ra bởi những sản phẩm oxy hóa glucose do chuỗi vận chuyển electron trong ty thể (mitochondrial electron transport chain).
Tăng glucose trong tế bào đẩy electron donors vào ty thể nhiều hơn, làm tăng sản xuất superoxide. Đứt sợi DNA do superoxide này làm hoạt hóa enzyme sửa chữa (repair enzyme) poly-(ADP-ribose) polymerase (PARP), là enzyme gây nên polymer hóa (polymerization) của ADP ribose (on) và sau đó ức chế (of) glyceraldehyde phosphate dehydrogenase (GAPDH) bởi polymers của ADP-ribose.
Khi hoạt hóa GAPDH bị giảm, đường chuyển hóa phân hủy glucose tăng lên theo đường ngược trở lại, đẩy chúng vào các đường khác như polyol, hexosamine, protein kinase C, AGEs. Tất cả những đường chuyển hóa bất lợi này sẽ gây nên nhiều biến chứng trong bệnh ĐTĐ.
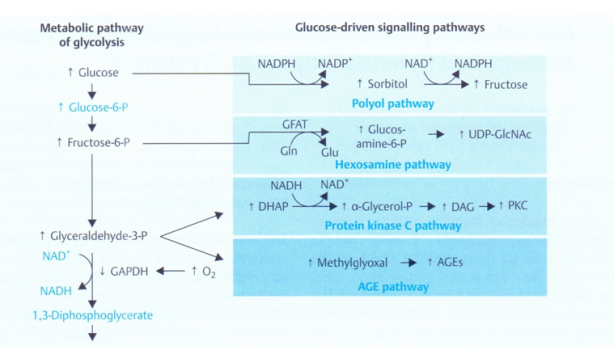
Bốn cơ chế chính, độc lập về tổn thương tổ chức do tăng glucose máu (Brownlee 2001,2005). Các đường chuyển hóa polyol, hexosamin, protein kinase C và AGE.
Trước hết, tăng glucose máu đưa đến sản xuất superoxide, superoxide làm giảm hoạt tính GAPDH sẽ chuyển ngược đường chuyển hóa glucose. Các chữ viết tắt: NAD, NADH= nicotinamide adenine dinucleotide và thể khử của nó. NADP, NADPH= nicotinamide adenine dinucleotide phosphate và thể khử của nó. GAPDH= glyceraldehyde phosphate dehydrogenase. Gln=glutamine, Glu= glucosamine. GFAT=glutamine:fructose-6-phosphate amidotransferase. DHAP= dyhydroxyacetonephosphate. DAG=diacylglycerol. PKC= protein kinase C. AGE=advanced glycation endproducts. UPD-GLcNSc=uridine diphossphate N-acetylglucosamine.
Glycat hóa protein không enzyme (Nonenzymatic protein glycation).
Tăng glucose máu đóng vai trò chủ yếu trong cơ chế bệnh sinh bệnh mạch máu do đái tháo đường. Tác dụng hủy hoại của nó thông qua các đường chuyển hóa khác nhau, non-enzymatic protein glycation là một trong những đường chuyển hóa đó. Glycat hóa protein không enzyme tạo ra các sản phẩm cuối glycat hóa bực cao (advanced glycation endproducts- AGEs) và các tiền chất của nó như methylglyoxal.
Methylglyoxal là một tác nhân quan trọng của glycat hóa tương tác với các nhóm amino tự do của glysine và arginine dư của proteins tạo nên AGEs trong tế bào gây nên biến đổi phiên mã gene (gene trancription), các protein matrix ngoài tế bào và các protein lưu hành trong máu và các bệnh lý mạch máu. (9)
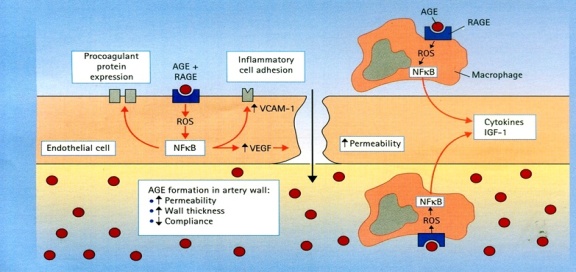
Cơ chế tăng tổn thương tế bào do AGEs + RAGE trong tế bào nội mạc và macrophages (8)
Trên bề mặt các tế bào nội mạc có các receptor đặc hiệu của AGE (RAGE), RAGE gắn với AGEs (4) là nguyên nhân làm tăng stress oxy hóa, tăng sản xuất reactive oxygen species (ROS), cytokins (TNFα. IL-1), các yếu tố phát triển (IGF-1, TGFβ ), các phân tử kết dính (adhesion molecules), và hoạt hóa chất làm tăng phiên mã của chuỗi nhẹ yếu tố nhân kappa (transcription nuclear factor kappa-light chain-enhancer).
Nhiều nghiên cứu đã nhấn mạnh vai trò đặc biệt của AGE như là một yếu tố trung gian trong cơ chế bệnh sinh bệnh ĐTĐ và các biến chứng của nó (5). Quá trình glycat hóa có thể ảnh hưởng đến vật liệu di truyền, đưa đến những thay đổi về bộc lộ gene.
Hoạt hóa đường chuyển hóa polyol.
Khi tăng glucose ở khoang ngoài tế bào, đường chuyển hóa glucose thứ hai cũng được hoạt hóa. Aldose reductase bình thường làm giảm aldehyde độc để bất hoạt alcohols.
Khi nồng độ glucose trong tế bào tăng, glucose được aldoreductase chuyển thành sorbitol, bằng cách đó làm giảm NADPH, một co-factor chủ yếu để phục hồi gluthation đã bị giảm. Gluthation là một chất chống oxy hóa (antioxidant) quan trọng trong tế bào, nếu như nồng độ chất này bị giảm, các tế bào dễ bị tổn thương do stress oxy hóa.
Chuyển glucose thành sorbitol và tích lũy chúng, nếu quá nhiều sẽ làm phồng (swelling) và tổn thương tổ chức. Tăng sorbitol đi kèm với những giọt myoinositol trong tế bào, có vai trò chuyển nạp (transduction) tín hiệu trong tế bào và điều hòa hoạt tính Na+/K+ATPase.
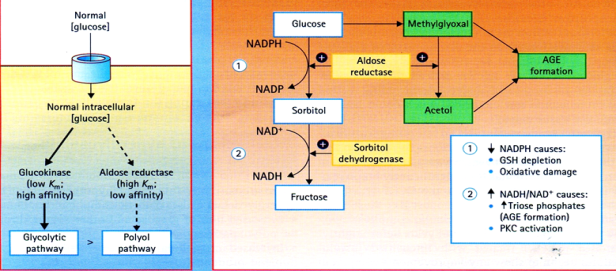
Tăng hoạt tính đường chuyển hóa polyol có vị trí chính ở võng mạc, đồng tử (pupil), thần kinh ngoại vi và tiểu cầu thận, nghĩa là những vùng mà vận chuyển glucose phụ thuộc insulin. Tích trữ sorbitol kích hoạt chuỗi các phản ứng đưa đến giảm hoạt tính Na/K ATPase, đôi khi đó là nguyên nhân gây nên rối loạn cấu trúc và chức năng các
cơ quan.
The polyol pathway. Trái. Đường chuyển hóa không hoạt hóa bình thường, nhưng sẽ trở nên hoạt hóa khi nồng độ glucose trong tế bào tăng lên. Phải. Hậu quả của glucose tăng chuyển hóa theo đường polyol, chuyển glucose để tạo glucose glycats mạnh (methylglyoxal, acetol và triose phosphates) làm tăng tổn thương do oxy hóa và tăng hoạt hóa PKC. GSH, reduced glutathione, NAD, nicotinamide adenine dinucleotide (8)
Đường chuyển hóa protein kinase C.
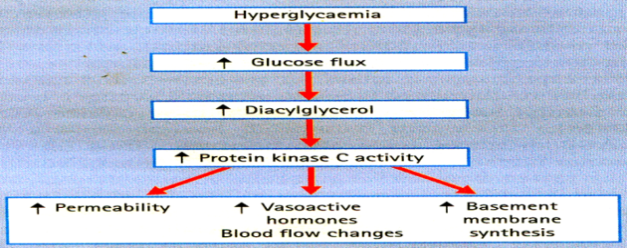
Được hoạt hóa khi tăng glucose máu. Tăng glucose trong tế bào làm tăng tổng hợp diacylglycerol, là một co-factor hoạt hóa protein kinase isoforms kinh điển.
Hoạt hóa protein kinase C làm hoạt hóa nuclear factor kappa B (NFkB), giảm nitric oxide synthase nội mạc (eNOS), tăng endothelin-1, Transforming growth-factor β (TGF-β), cũng như plasminogen activator inhibitor-1 (PAI-1), gây nên những bất thường dòng máu, làm tặc mạch.
Trong các tế bào sự bộc lộ aldose reductase thấp hoặc không có, tăng glucose máu làm tăng sản xuất diacyloglycerol, nó làm tăng hoạt tính protein kinase C.
Hoạt hóa mạnh protein kinae C đưa đến các rối loạn cấu trúc và chức năng trong tế bào. Tăng glucose máu cũng có thể có tác dụng đối nghịch đối với protein kinase C trong những typ tế bào khác nhau.
Ví dụ, tế bào perycyte võng mạc, các tế bào này bộc lộ cao aldoreductase trong tình trạng tăng glucose máu bắt đầu atrophy, là nguyên nhân gây nên tình trạng phình vi mạch, mặt khác, các tế bào nội mạc, vì không có enzyme trong suốt quá trình tăng sinh đưa đến bịt tắc lòng mạch (7).
Đường chuyển hóa hexosamine.
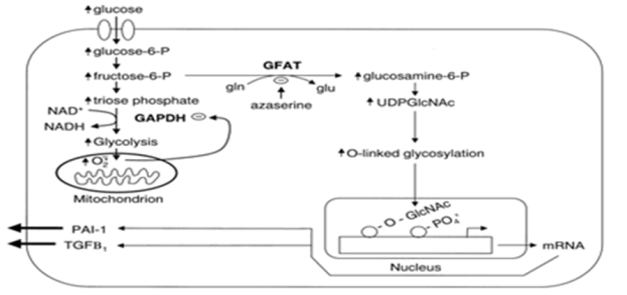
Sản xuất quá mức superoxide trong ty thể do tăng glucose máu hoạt hóa đường chuyển hóa hexosamine và tăng bộc lộ gene phụ thuộc Sp1.
Tăng phân hủy glucose sẽ tạo ra superoxide bởi tăng chệnh hóa điện tử proton xẩy ra do chuỗi vận chuyển electron trong ty thể. Superoxide khi đã được tạo ra sẽ ức chế hoạt tính enzyme GAPDH, và chuyển fructose-6-phosphate vào đường chuyển hóa hexosamine, làm tăng UPD-GlcNAc. Tăng này sẽ làm tăng O-GlcNAcylation của Sp1, tăng giao dịch (transactivation) và bộc lộ gene phụ thuộc Sp1.
Enzyme Glutamine:fructose 6- phosphate amidotransferase (GFAT) chuyển ngược fructose 6 – phosphate thành glucosamine 6- phosphate, cuối cùng thành uridine diphossphate N- acetylglucosamine (UDPGIcNAc), sản phẩm chuyển hóa cuối này làm biến đổi các yếu tố phiên mã, làm tăng bộc lộ PAI-1. Và TGF-β, do đó làm biến đổi các phân tử tín hiệu (signalling molecules) là tăng tình trạng kháng insulin.
Rối loạn chuyển hóa Proteoglycans.
Trong quá trình phát triển bệnh mạch máu do ĐTĐ, những rối loạn chuyển hóa proteoglycans có vai trò quan trọng. Glycosaminoglycans (GAG) là một phần chính trong cơ chế bệnh sinh bệnh thận
do ĐTĐ.
Proteoglycans là những thành phần phân tử lớn của matrix ngoài tế bào, chúng gồm lõi protein gắn hóa trị hai với các chuỗi glycosaminoglycan giai đoạn cao của quá trình phân chia.
Tăng glucose máu làm rối loạn chuyển hóa của proteoglycan như heparan sulfate. Thiếu heparan sulfate (HS) đưa đến xơ tiểu cầu thận và làm tăng nhanh những thay đổi do vữa xơ trong vi mạch. Thiếu GAG làm tăng tăng sinh tế bào cơ trơn.
Tổn thương mạch máu lớn: vai trò của acid béo tự do (free fatty acid-FFA)
Tư liệu từ UKPDS (1998) đã chứng minh rằng, tăng glucose máu không phải là yếu tố quyết định chính biến chứng bệnh mạch máu lớn do ĐTĐ, trong khi tăng gần 10 lần các biến cố vi mạch khi HbA1c tăng từ 5,5 đến 9,5%, nguy cơ bệnh mạch máu lớn chỉ tăng 2 lần. The San Antonio Heart Study cũng đã chứng minh, kháng insulin là yếu tố chính nguy cơ tim mạch.
Hanson et al (2002) đã nghiên cứu ở những người có dung nạp glucose bình thường đã phát hiện thấy nguy cơ các biến cố tim mạch tăng ở những người có kháng insulin rõ, ngay cả sau khi điều chỉnh với 7 yếu tố nguy cơ tim mạch đã biết (như lipid, huyết áp, hút thuốc…).
Những dấu hiệu này nhấn mạnh về vai trò rõ rệt về hậu quả của kháng insulin khác nhau giữa những yếu tố đã được ghi nhận
trước đây.
Để làm sáng tỏ cơ chế liên quan đến các biến chứng mạch máu lớn, các tế bào nội mạc động mạch người nuôi cấy, và mô hình thực nghiệm trên động vật đã được tiến hành nghiên cứu (Du et al 2006).
Khi tín hiệu insulin bị rối loạn trong tế bào mỡ, có hiện tượng tăng vận chuyển các acid béo tự do (FFAs) từ tế bào mỡ (adipocyte) vào tế bào nội mạc động mạch.
Khi tín hiệu insulin bị rối loạn trong tế bào nội mạc mạch máu, các FFAs này bị oxy hóa trong ty thể cung cấp những donors điện tử giống nhau đến chuỗi vận chuyển electron trong ty thể như oxy hóa glucose, vì vậy, sản xuất quá nhiều ROS do FFA sẽ hoạt hóa các đường rối loạnchuyển hóatương tự như đã chứng minh khi tăng glucose máu. Tăng sản xuất
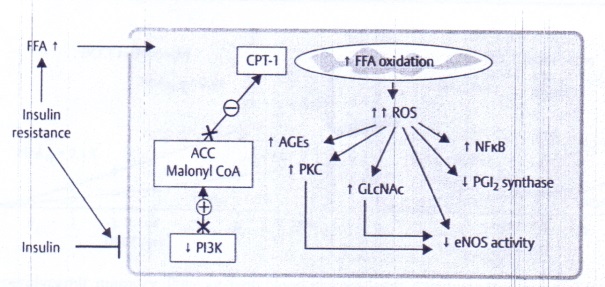
Tế bào nội mạc động mạch
Các acid béo tự do và cơ chế gây tổn thương do tăng glucose máu.
Kháng insulin làm tăng oxy hóa acid béo tự do trong tế bào nội mạc động mạch, hoạt hóa các tín hiệu tiền vữa xơ (proatherogenic signals) và ức chế các enzymes chống vữa xơ (Du et al 2006) . FFA = free fatty acids, CPT-1 = carnitine palmitoyltransferase-1, ACC = acetyl-CoA carboxylase.GlcNAc = N-acetylglucosamine, PGI2 = Prostacyclin, PI3K = Phosphoinositide 3-kinase.
Superoxide từ tăng oxy hóa FFAs cũng do bất hoạt trực tiếp oxy hóa prostacylin synthase và eNOS, hai enzymes chống vữa xơ đặc biệt này bị giảm ở BN ĐTĐ.
Hai mô hình nghiên cứu trên động vật không bị ĐTĐ, tình trạng kháng insulin được xác định băng di truyền hoặc gây bằng chế độ ăn giàu mỡ. Sự bất hoạt một số enzymes được ngăn chặn bằng ức chế giải phóng FFA từ tế bào mỡ bằng antipolytic nocotinic acid, bằng ngừng sản xuất quá mức superoxide trong ty thể, và bằng hạn chế oxy hóa FFA bằng etoximir và các yếu tố ức chế carnitine palmitoyltransferase-1 (CPT-1).
Những cơ chế bổ sung này về tổn thương tổ chức và mạch máu tham gia thúc đẩy bệnh VXĐM và tăng nguy cơ bệnh mạch máu ở BN ĐTĐ, cũng như làm tăng tình trạng kháng insulin nói riêng.
Ảnh hưởng lâu dài trí nhớ tăng
glucose máu.
Nhiều nghiên cứu ngẫu nhiên đã xác minh rằng, kiểm soát glucose máu sớm, tích cực làm giảm nguy cơ các biến chứng do bệnh ĐTĐ gây nên. Một số công trình khác ủng hộ nhận xét ảnh hưởng lâu dài của kiểm soát chuyển hóa sớm đối với các kết cục lâm sàng. Để giải thích cho những nhận xét trên đây, nhiều ý kiến cho rằng liên quan đến “ trí nhớ tăng glucose máu- hyperglycemic memory”, và gần đây đang nói nhiều “trí nhớ chuyển hóa (metabolic memory”) (Ihnat et
al 2007).
Bằng chứng đầu tiên về trí nhớ tăng glucose máu ở người xuất phát từ kết quả nghiên cứu trong thời gian dài của hai công trình ở BN ĐTĐ typ 1: DCCT (1993) “Diabetes control and complications trial”, và sau đó là EDIC (Epidemiology of diabetes interventions and complications (JAMA 2002). DCCT/EDIC nghiên cứu bao gồm 1441 người ở độ tuổi từ 13- 39, được chẩn đoán ĐTĐ typ 1, điều trị tích cực bằng insulin hoặc truyền thống.
Sau thời gian theo dõi trung bình 6,5 năm, bệnh võng mạc tiến triển ở cả hai nhóm, nhưng độ dốc ở nhóm điều trị truyền thống dốc hơn, tương quan có ý nghĩa với nồng độ HbA1c: 9,1% nhóm điều trị truyền thống và 7,3% nhóm điều trị tích cực.
Sau giai đoạn nghiên cứu so sánh đối chứng ngẫu nhiên của DCCT kết thúc, những người đã tham gia nghiên cứu theo phương pháp điều trị tích cực động viên tiếp tục. Những người tham gia khi bắt đầu ở nhóm điều trị truyền thống khuyên chuyển sang điều trị theo chế độ tích cực. Kết quả là tăng HbA1c nhóm vẫn điều trị tích cực từ 7,3% lên 7,9%, và từ 9,1% (ở nhóm trước đây điều trị truyền thống chuyển sang điều trị tích cực) xuống 8,3%.
Sự khác biệt giá trị HbA1c giống nhau có ý nghĩa thống kê và duy trì trong suốt thời gian nghiên cưú EDIC, nhưng đây là một quan sát rất quan trọng : độ dốc tiến triển bệnh võng mạc đã không thay đổi ở cả hai nhóm, độ dốc hai đường biểu diễn giống nhau, hiện tượng này chứng tỏ rằng nồng độ glucose cao hơn trước đó quyết định một cách chắc chắn nguy cơ các biến chứng do ĐTĐ trong tương lai, ngay những năm sau đó, nồng độ glucose máu thấp hơn cũng không làm thay đổi tốc độ phát triển các biến chứng.
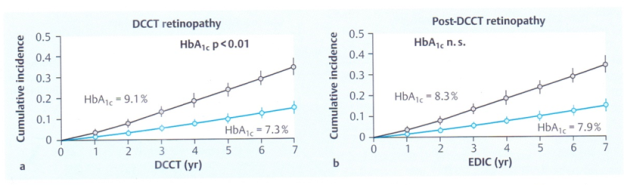
Trí nhớ tăng glucose máu. Nồng độ glucose cao trước đây quyết định mức độ các tổn thương trong tương lai các biến chứng bệnh ĐTĐ. Độ dốc đường biểu diễn tỷ lệ cộng dồn các biến chứng không thay đổi, mặc dầu những năm sau đó nồng độ glucose máu thấp hơn. a: Tỷ lệ cộng dồn bệnh võng mạc do ĐTĐ trong nghiên cứu DCCT.1993. b: Tỷ lệ bệnh cõng mạc theo dõi về sau trong nghiên cứu EDIC (JAMA 2002)
Hiện tượng này cũng xẩy ra ở Bn ĐTĐ typ 2, ở trường hợp này gọi là “ trí nhớ tăng glucose máu” (hyperglycemic memory), có thể quan trọng hơn khi xét đến bệnh ĐTĐ typ 2 bị chẩn đoán muộn trong thời gian dài. Hầu hết BN ĐTĐ typ 2 được chẩn đoán sau khi họ đã bị bệnh sau 8-10 năm, một thời gian dài tồn tại tăng glucose máu ở những mức độ khác nhau.
Cơ chế sinh lý bệnh có thể về trí nhớ tăng glucose máu được nghiên cứu khi dùng tế bào nội mạc động mạch của người và mô hình thực nghiệm trên động vật loài gậm nhấm thỏ không bị ĐTĐ (EL-Osta et al. 2008).
Cả in vitro và in vitro, tăng glucose máu nhất thời (transient) (16h trong thực nhiệm in vitro và 6h trong in vivo) hoạt hóa những thay đổi epigenetics (biểu sinh) trong thời gian dài ở vùng khởi động (promoter) của NFkB tiểu đơn vị 65. Những thay đổi này kéo dài trong 6 ngày sau khi glucose máu bình thường, và sẽ bị ngăn lại bằng làm giảm tạo thành ROS hoặc tạo thành methylglyoxal.
Những tác dụng epigenic kéo dài muộn về sau có sự trung gian bởi tập hợp các enzymes tham gia tái biến đổi histone (histone modifying enzymes), các enzymes này tái cấu trúc các sợi chromatin DNA, nhờ vậy quá trình phiên mã trong gene xẩy ra. (Turner -2007).
Khái niệm về “Trí nhớ tăng glucose máu” rất có giá trị trong thực hành lâm sàng. Một mặt phải điều trị sớm để đưa glucose màu trở về bình thường, mặt khác , bổ sung các thuốc có tác dụng làm giảm ROS và glycat hóa xẩy ra trong tế bào để hạn chế tới mức thấp nhất hoặc có thể đảo ngược các biến chứng mạn tính BN ĐTĐ.
TÀI LIỆU THAM KHẢO
- The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and neph-ropathy in patients with type 1 diabetes four years after a trial of inten-sive therapy. N Engl J Med 2000; 342: 381–389.
- Writing Team for the Diabetes Control and Complications Trial/Epide-miology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002; 287: 2563–2569.
- Ihnat MA, Thorpe JE, Ceriello A. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabet Med 2007; 24: 582–586.
- Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010; 59: 249–255.
- Yan SF, Ramasamy R, Schmidt AM. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab 2008; 4: 285–293.
- De Boer IH, Kestenbaum B, Rue TC et al.; Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Study Research Group.Insulin therapy, hyperglycemia, and hypertension in type 1 diabetes mellitus. Arch Intern Med 2008; 168: 1867–1873
- Kowluru RA, Zhong Q, Kanwar M. Metabolic memory and diabetic retinopathy: role of inflammatory mediators in retinal pericytes. Exp Eye Res 2010; 90: 617–623.
- Tetsuya Taguchi and Michael Brownlee. Textbook of diabetes 2005. Chapter 47.1-17
- P.J.Thornalley and P. Kempler. Complications of diabetes mellitus: Pathophysiology and Pathogenetically-based treatment options. International Expert workshop. September 2008. Rome, Italy. 1-7
- Gerrits EG, Lutgers HL, Kleefstra N, Groenier KH, Smit AJ, Gans RO, Bilo HJ. Skin advanced glycation end product accumulation is poorly reflected by glycemic control in type 2 diabetic patients (zodiac-9). J Diabetes Sci Technol, 2008, 2: 572–577
- Gradinaru D, Borsa C, Ionescu C, Margina D. Advanced oxidative and glycoxidative protein damage markers in the elderly with type 2 diabetes. J Proteomics, 2013, 92: 313–322
- Guay C, Regazzi R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat Rev Endocrinol, 2013, 9: 513–521
- Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T. Endothelial microparticle-mediated transfer of microRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damagedendothelial microparticles. Circulation, 2013, 128: 2026–2038
